
 下载APP
下载APP 报料
报料 关于
关于
湖南省妇幼保健院医学遗传科博士 刘静 大众卫生报 2022-08-05 16:53:28
小明是个刚出生不久的宝宝,家人正沉浸在迎接新生命的喜悦中时,妈妈却发现小明吃奶总是需要很长时间,吃的不多还很费劲,经常有呛咳,反复呼吸道感染、三个月了还抬头不稳,浑身软软的。
妈妈焦虑地抱着小明前来湖南省妇幼保健院遗传门诊咨询,经过一系列医学检查,生化检查显示正常,肌电图提示神经源性损害,临床诊断考虑为“脊髓性肌萎缩症”,随后的遗传学检测提示其SMN1基因第7号外显子纯合缺失,为常染色体隐性遗传性疾病,可能小明的爸妈都是疾病的携带者。
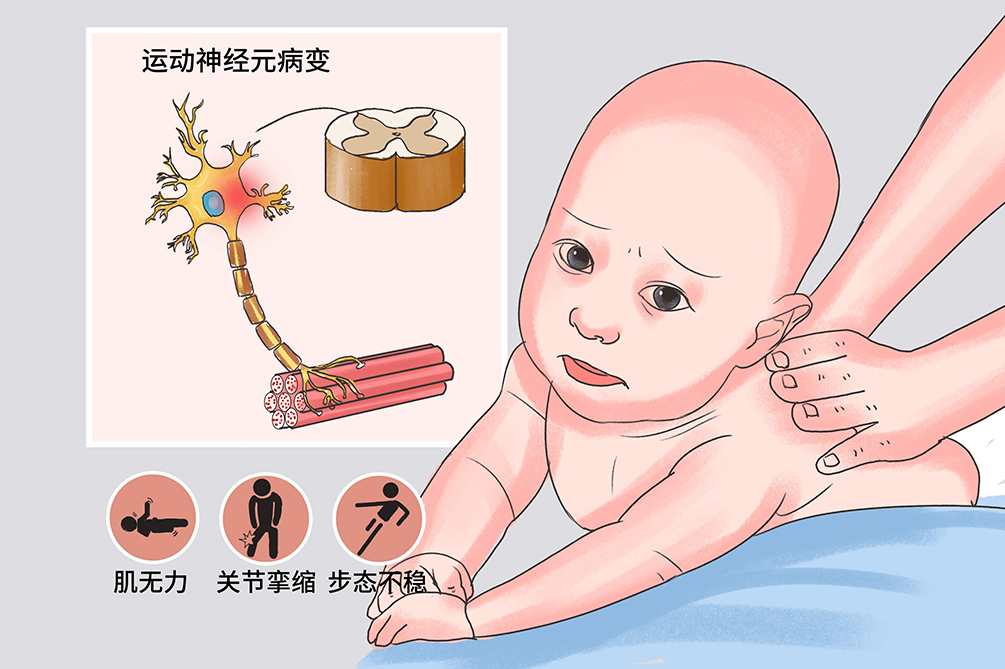
说起这类罕见病,大家可能会非常陌生,2021年一段网络爆火的视频让大家认识了这样一个常见的“罕见病”-脊髓性肌萎缩症,国家医保局经过8轮协商谈判将70万元一针的治疗脊髓性肌萎缩症(SMA)的药物“诺西那生钠”砍价到了33000元,成功纳入到了医保药品体系之中,为SMA罕见病患儿家庭带来了治疗的希望与可能。
脊髓性肌萎缩症(SMA),是一类由脊髓前角运动神经元变性导致肌无力、肌萎缩的疾病,是一种常染色体单基因遗传病,呈隐性遗传模式,我国发病率约为1/6000~1/10000,男女发病率无显著差异。根据患者起病年龄和临床病程,将SMA由重到轻分为4型,1型6个月内起病,从来不能独坐,2岁以内死亡;2型6-18个月起病,从来不能独站,平均寿命25岁;3型幼儿期起病,能独站,独走;4型成年期起病。这个疾病的共同特点是脊髓前角细胞变性,神经元细胞和肌肉组织就像是指挥官和士兵的关系,神经元细胞变性之后失去了指挥官的指示,其下游控制的肌肉组织就开始不工作了,最终导致的是进行性、对称性,肢体近端为主的广泛性弛缓性麻痹与肌萎缩,但是患者的智力发育及感觉均正常,所以SMA患者是真的没有力气活动,而不是因为偷懒不想动。
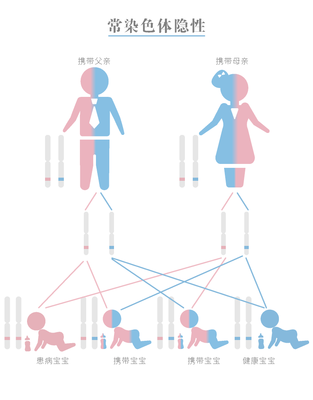
SMA是常染色体隐性遗传性疾病,致病基因SMN1位于7号染色体上,如果仅有一个致病突变,我们称为携带者,自身没有临床症状。若夫妻双方均携带同一个致病基因,生育后代的过程中,有可能各自将一半的有害的基因传递给后代,后代有25%的可能遗传到父母双方的致病突变,成为真正的“患者”。针对SMA患者,早诊、早治、早康复是提高生活质量的三大关键要素。因此,我们提倡在孕前或者孕早期对夫妻双方进行SMA携带者筛查,通过早期的筛查,提前发现潜在的“携带者”,发现夫妻同为携带者时,我们可以通过胚胎植入前诊断或者介入行产前诊断来干预,提前诊断胚胎或者胎儿是否罹患SMA,避免悲剧发生。
湖南省妇幼保健院医学遗传科目前已开展扩展性携带者筛查以及遗传性出生缺陷产前诊断等,如果有遗传方面的问题,欢迎进行咨询、检查。
湖南省妇幼保健院医学遗传科博士 刘静
一审:王璐
二审:梁湘茂
三审;汤江峰

责编:王璐
来源:大众卫生报
我要问
 湘公网安备 43010502000374号
湘公网安备 43010502000374号